Alternative Splicing (rMATS) Differential Analysis Document
Introduction
TIP
Alternative Splicing (AS) is a key mechanism in eukaryotes that regulates gene expression and generates protein diversity. Through different splicing patterns, a single gene can produce multiple mRNA isoforms, which can then translate into proteins with distinct functions. rMATS (replicate Multivariate Analysis of Transcript Splicing) is a powerful and flexible computational tool specifically designed to detect differential alternative splicing events from RNA-Seq data.
In single-cell sequencing data analysis, rMATS can help researchers compare alternative splicing patterns between different cell populations (such as different cell types or treatment groups), identifying key splicing changes associated with specific biological processes or disease states. Compared to differential gene expression analysis, differential alternative splicing analysis provides another dimension of gene regulation information, helping to reveal the molecular basis of cellular heterogeneity and functional diversity in greater depth.
This document aims to provide researchers with a comprehensive technical guide for rMATS analysis, covering its basic principles, operation methods on the SeekSoul™ Online, result interpretation, and frequently asked questions, helping you quickly master and apply this tool for in-depth data mining.
rMATS Theoretical Foundation
Core Principles
rMATS quantifies and compares the frequency of specific alternative splicing events between different sample groups by analyzing RNA-Seq reads aligned to transcripts. It counts the number of reads supporting "exon inclusion" and "exon skipping" and uses multivariate statistical models to evaluate whether differences in "Percent Spliced In" (PSI or IncLevel) between two groups of samples are significant.
Five Types of Alternative Splicing Events Identified by rMATS
rMATS can identify and analyze the following five most common types of alternative splicing events:
- SE (Skipped Exon):An exon is included in some transcripts and skipped in others. This is the most common and well-studied type of alternative splicing.
- A5SS (Alternative 5' Splice Site):The 3' splice site of an exon is fixed, but different 5' splice sites are used, resulting in changes to exon length.
- A3SS (Alternative 3' Splice Site):In contrast to A5SS, the 5' splice site is fixed, but different 3' splice sites are used.
- MXE (Mutually Exclusive Exons):Two or more exons can only appear in one form in transcripts, with one excluding the others.
- RI (Retained Intron):An intron is normally spliced out in some transcripts but retained in others, becoming part of the mature mRNA.

(Image source: rMATS official website)
SeekSoul™ Online Operation Guide
On the SeekSoul™ Online, the rMATS analysis workflow is designed to be intuitive and user-friendly. You don't need to write code; you can complete the analysis simply through the parameter configuration interface.
Preparation Before Analysis
TIP
rMATS analysis relies on high-quality BAM alignment files. Before starting the analysis, please ensure:
- Data has been aligned: Your single-cell data has been aligned and BAM files have been generated.
- Appropriate grouping has been selected: Differential alternative splicing analysis needs to be performed between two or more clearly defined biological groups (e.g., treatment group vs. control group, disease samples vs. healthy samples, different cell types, etc.).
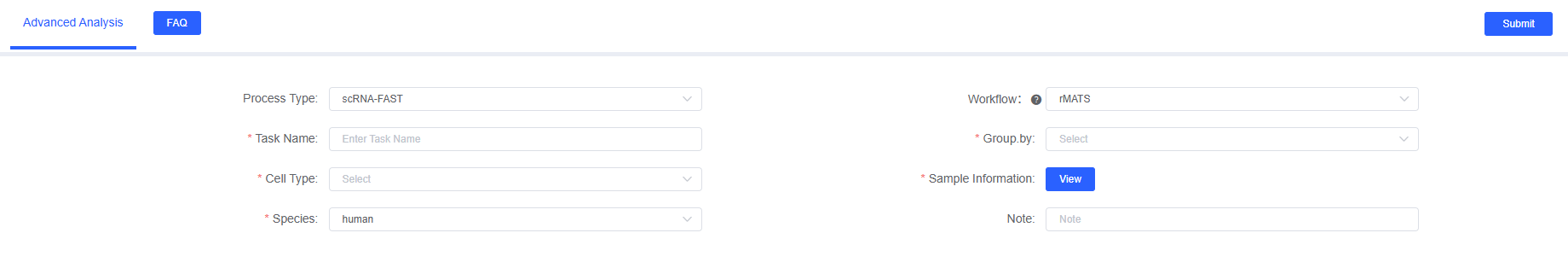
Parameter Details
The following table details the main parameters and their explanations for the rMATS analysis module on the SeekSoul™ Online.
| Interface Parameter | Description |
|---|---|
| Task Name | The name of this analysis task, must start with an English letter, and can contain English letters, numbers, underscores, and Chinese characters. |
| Grouping.by | The specified metadata name used to define comparison groups, such as celltype or Group. |
| Cell Type | Select the cell type or group to be analyzed. The platform will perform group comparisons based on this. |
| Sample Information | Select the samples to be analyzed and specify the corresponding BAM file paths. |
| Species | Select the species corresponding to your data; options include human, mouse, or fruit fly. |
| Notes | Custom notes for recording analysis details. |
Operation Workflow
- Enter the analysis module: Navigate to the "Advanced Analysis" module on the SeekSoul™ Online and select "rMATS Alternative Splicing".
- Create a new task: Name your analysis task.
- Configure parameters: Set the task name, grouping factor, cell type, BAM file paths, etc., according to the above guidelines.
- Submit the task: After confirming the parameters are correct, click the "Submit" button and wait for the analysis to complete.
- Download and view: After the analysis is complete, download and view the generated analysis report and result files in the task list.
Result Interpretation
The rMATS analysis report includes statistical charts and detailed data tables. The following is a detailed interpretation of the core results.
Alternative Splicing Event Statistics
The report first presents statistics on the number of five types of alternative splicing events detected in different comparison groups.
- Chart Interpretation: This bar chart shows the total number of five types of alternative splicing events (SE, MXE, A5SS, A3SS, RI) detected in a comparison group (e.g.,
clusterDCsvs. all other cells). - Analysis Points:
- Quickly understand the overall situation of alternative splicing events in the data.
- Typically, SE (Skipped Exon) events are the most numerous, while RI (Retained Intron) events are relatively fewer.
Differential Alternative Splicing Event List
This is the core result of rMATS analysis, listing all detected differential alternative splicing events in table form. The report typically displays the result file for SE (Skipped Exon) events by default.
- File Interpretation (
*.MATS.JC.txt):- GeneSymbol: The name of the gene where the event is located.
- chr, strand, ExonStart_0base, ExonEnd: Chromosomal position information of the event.
- IJC_SAMPLE_1, SJC_SAMPLE_1: Number of reads supporting "inclusion" (Inclusion Junction Count) and "skipping" (Skipping Junction Count) in Group 1.
- IJC_SAMPLE_2, SJC_SAMPLE_2: Number of reads supporting "inclusion" and "skipping" in Group 2.
- IncLevel1, IncLevel2: "Inclusion Level" for Group 1 and Group 2, calculated as
(IJC / IncFormLen) / ((IJC / IncFormLen) + (SJC / SkipFormLen)). This value ranges from 0 to 1, with values closer to 1 indicating a higher tendency for the exon to be included. - IncLevelDifference: The difference between
IncLevel1andIncLevel2. This is a key indicator of the magnitude of the differential effect; a larger absolute value indicates a more significant difference. - PValue, FDR: The p-value for statistical significance and the FDR value after multiple test correction. Typically,
FDR < 0.05and|IncLevelDifference| > 0.1are used as criteria for screening significant differential events.
Notes
- Result Validation: The results of rMATS are based on computational inference. For key differential alternative splicing events, it is strongly recommended to validate them using molecular biology experiments such as RT-PCR.
- Screening Criteria: When screening for significant differential events, don't just look at the
FDRvalue. Be sure to combine it withIncLevelDifferenceto evaluate the biological significance of the difference. An event with a very smallFDRbut anIncLevelDifferenceof only 0.01 may have negligible biological effects. - Sample Quality: The sequencing depth and read length of RNA-Seq will affect the ability to detect alternative splicing events. Greater depth and longer read lengths (especially paired-end sequencing) are more conducive to accurately identifying and quantifying splicing events.
- Combination with Differential Gene Expression: Combining differential alternative splicing analysis with differential gene expression analysis can provide a more comprehensive understanding of gene regulatory changes at the transcript and protein levels. Sometimes, a gene's overall expression level remains unchanged, but the proportion of its isoforms changes dramatically, which can also have important biological functions.
Frequently Asked Questions (FAQ)
Q1: What does "IncLevel" (Inclusion Level) mean in the rMATS analysis report?
A: "IncLevel" (or PSI, Percent Spliced In) is an indicator that measures the frequency at which an alternative exon is included in mature mRNA. Its value ranges from 0 to 1. For example, an SE event with an IncLevel of 0.8 means that in that sample, 80% of transcripts include this exon, while 20% skip it. IncLevelDifference compares this frequency difference between two groups of samples and is a core indicator for judging the magnitude of differential alternative splicing effects.
Q2: How should I screen for meaningful differential alternative splicing events?
A: A commonly used screening criterion is FDR < 0.05 and |IncLevelDifference| > 0.1.
FDR < 0.05ensures statistical significance.|IncLevelDifference| > 0.1ensures that the difference has a certain biological effect size (i.e., at least a 10% change in exon inclusion proportion between the two groups). You can adjust theIncLevelDifferencethreshold according to research needs, for example, using 0.2 or higher to obtain stronger candidate events.
Q3: Why are there many alternative splicing events for a single gene in my analysis results?
A: This is normal. A gene, especially a large gene with a complex structure, may contain multiple alternative exons or alternative splice sites. Therefore, rMATS may detect multiple independent SE, A5SS, or RI events on the same gene. During analysis, you can examine these events one by one, or focus on the events with the most significant differences.
Q4: What is the difference between rMATS and finding differentially expressed genes? Which one should I do?
A: They analyze gene regulation from different perspectives, and it's best to do both.
- Differential gene expression analysis focuses on the "quantitative change" of genes: whether the overall transcription level of a gene is upregulated or downregulated.
- Differential alternative splicing analysis (rMATS) focuses on the "qualitative change" of genes: whether the proportion of different mRNA isoforms produced by a gene changes, even if the overall gene expression level remains unchanged. A gene may show no difference in expression level, but its main functional isoform may change from A to B, which can also cause important functional changes. Therefore, combining both analyses provides a more comprehensive perspective.
References
Wang Y, Xie Z, Kutschera E, Adams JI, Kadash-Edmondson KE, Xing Y. rMATS-turbo: an efficient and flexible computational tool for alternative splicing analysis of large-scale RNA-seq data. Nature Protocols, 2024. doi: 10.1038/s41596-023-00944-2.
Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, Xing Y. rMATS: Robust and Flexible Detection of Differential Alternative Splicing from Replicate RNA-Seq Data. PNAS, 2014; 111(51):E5593-601. doi: 10.1073/pnas.1419161111.
Park JW, Tokheim C, Shen S, Xing Y. Identifying differential alternative splicing events from RNA sequencing data using RNASeq-MATS. Methods in Molecular Biology: Deep Sequencing Data Analysis, 2013; 1038:171-179. doi: 10.1007/978-1-62703-514-9_10.
Shen S, Park JW, Huang J, Dittmar KA, Lu ZX, Zhou Q, Carstens RP, Xing Y. MATS: A Bayesian Framework for Flexible Detection of Differential Alternative Splicing from RNA-Seq Data. Nucleic Acids Research, 2012; 40(8):e61. doi: 10.1093/nar/gkr1291.